LA JOLLA, CA—Members of the TET family of proteins help protect against cancer by regulating the chemical state of DNA —and thus turning growth-promoting genes on or off. The latest findings reported by researchers at La Jolla Institute for Immunology illustrate just how important TET proteins are in controlling cell proliferation and cell fate.
For the study, published in the December 20, 2016, edition of Nature Immunology, Anjana Rao, PhD, a professor at the La Jolla Institute, genetically engineered mice to lack both TET2 and TET3 in T cells. The mice developed a lethal disease resembling lymphoma within weeks of birth, their spleens and livers bloated with iNKT cells, a normally rare kind of T cell. This finding recapitulated the features of many human blood cancers, including those involving T cells, in which TET2 is often mutated or lost.
“We knew that TET proteins were involved in human cancer but we didn’t know how they regulated T cell development,” says Angeliki Tsagaratou, Ph.D., an instructor in the Rao lab and the study’s first author. “In the new study we saw huge increases in the proliferation of the special iNKT cells in TET2/3 mutant mice. Once growth control was lost, those cells underwent the kind of malignant transformation that gives rise to T cell lymphoma in humans.” The results demonstrate how TET proteins serve as anti-cancer factors called tumor suppressors and suggest ways to block malignancy in cancers marked by TET mutations.
Members of the TET family of enzymes help rewrite the epigenome, the regulatory layer of chemical modifications that sits atop the genome and helps determine gene activity without changing the letters of DNA. In addition to the four letters or bases in DNA – A, C, G and T –
This base is formed from the DNA base cytosine (C) by addition of a methyl group, and so is called mC (m for methyl). The levels of mC are altered in cancer cells and during the development of embryos.
However, until the discovery of TET proteins in 2009 in the laboratory of Rao, then at Harvard Medical School, it was not known how mC could be converted back to regain C. Dr. Rao’s team showed that TET proteins were able to convert 5mC to a sixth base, known as 5hmC. 5hmC is indirectly converted back to C, thus restoring the status quo.
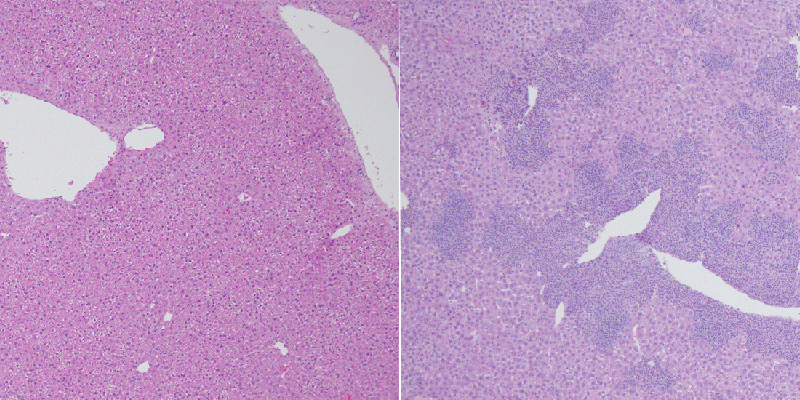
DNA modifications of this type in part govern how compressed and “expressible” a strand is: in general, DNA methylation coils up genes to silence them, while less methylated DNA strands, or strands that possess 5hmC, are more accessible and more likely to be expressed, which means they are directing the synthesis of a particular protein. “When TET proteins are lost, iNKT cells that lack them apparently become trapped in an immature, highly proliferative state,” explains Tsagaratou. “Unlike normal cells, they can’t switch off growth-promoting genes: they just keep dividing.”
DNA sequencing followed by bioinformatic data-crunching revealed the kinds of abnormal DNA methylation patterns typically seen after TET protein loss. That suggested that improper DNA modifications in the TET2/3 mutant T cells allowed unchecked expression of cancer- and inflammation-associated genes. Edahí González-Avalos, one of the two second authors of the study, conducted most of that analysis.
“Without computer analysis of sequencing results, we would not have been able to determine relationships between DNA methylation, how accessible regions of the genome were, global gene expression, or the emergence of cancer cells,” says
González-Avalos, a graduate student in UCSD’s Bioinformatics Graduate Program. “Without computational tools, this study could have taken many, many years!”
A critical test reported in the paper demonstrates how insidious even a few perpetually immature iNKT cells can be. In it, the team transferred a small number of iNKT cells lacking Tet2/3 from mutant mice into adult mice with a robust immune system. But even those mice soon developed lymphoproliferative disease as lethal as that seen in TET2/3 mutant mice.
“We weren’t expecting this,” says Tsagaratou. “When we transferred mutant cells we thought healthy mice would control their expansion. But in three months mutant cells took over the mouse’s immune system and rapidly gave rise to tumors.” The lesson of this story? That a functional immune system is no defense against malignancy once deregulated, pro-inflammatory iNKT cells gain a foothold.
In a 2015 Nature Communications paper, Rao, who heads LJI’s Division of Signaling and Gene Expression, reported that TET2/3 mutations caused myeloid disease resembling acute myeloid leukemia in mice. The new study extends these findings to a different class of hematological cancers, namely lymphoid cancer, which is caused by abnormal activity of immune T or B cells.
“Right now we don’t know how TET mutations specifically contribute to either T cell lymphomas or leukemias. But we think these mutations are early events in both,” says Tsagaratou. Thus the search is on is to discover additional cancer-causing genes “downstream” of TET mutations that drive uncontrolled cell division in either context. “Identification of additional factors would give us a broad idea of all steps in pathway and provide multiple targets to hit.”
In addition to Tsagaratou and González-Avalos, Sini Rautio of Aalto University School of Science, in Aalto, Finland, was a co-second author of the paper, contributing significantly to the bioinformatic analysis of genome-wide sequencing data. Also contributing were James Scott Browne, Ph.D., Susan Togher, and William A. Pastor, Ph.D., all from LJI; Ellen V. Rothenberg, Ph.D., of Caltech; and bioinformaticians Lukas Chavez Ph.D., of the German Cancer Research Center in Heidelberg and Harri Lähdesmäki, Ph.D., of Aalto University School of Science, the PhD thesis supervisor of Sini Rautio.
The study was funded by the NIH (R01 grants AI44432, CA151535 and R35CA210043); a Leukemia & Lymphoma Society grant (6187-12, to A.R.); and an Academy of Finland Centre of Excellence in Molecular Systems Immunology and Physiology Research grant (to H.L.). Other funding was from the Cancer Research Institute, the Academy of Finland Centre of Excellence in Molecular Systems, Immunology and Physiology Research program, the Damon Runyon Cancer Research Foundation (DRG-2069-11), and the National Science Foundation. doi:10.1038/ni.3630
About La Jolla Institute for Immunology
The La Jolla Institute for Immunology is dedicated to understanding the intricacies and power of the immune system so that we may apply that knowledge to promote human health and prevent a wide range of diseases. Since its founding in 1988 as an independent, nonprofit research organization, the Institute has made numerous advances leading toward its goal: life without disease.


